Basically, the concept behind Phylosift is to provide for high quality, automated, high throughput phylogeny-driven analysis of metagenomic sequence data. The software was developed openly on github and has been available in some form for more than a year. Aaron, Holly, Erick and I have discussed it extensively in various talks around the world and thus we assume some are already familiar with it.
This project was coordinated by Aaron Darling, who was a Project Scientist in my lab and is now a Professor at the University of Technology Sydney. Also involved were Holly Bik (post doc in the lab), Guillaume Jospin (Bioinformatics Engineer in the lab), Eric Lowe (was a UC Davis undergrad working in the lab) and Erick Matsen (from the FHCRC).
Abstract:
Like all organisms on the planet, environmental microbes are subject to the forces of molecular evolution. Metagenomic sequencing provides a means to access the DNA sequence of uncultured microbes. By combining DNA sequencing of microbial communities with evolutionary modeling and phylogenetic analysis we might obtain new insights into microbiology and also provide a basis for practical tools such as forensic pathogen detection.
In this work we present an approach to leverage phylogenetic analysis of metagenomic sequence data to conduct several types of analysis. First, we present a method to conduct phylogeny-driven Bayesian hypothesis tests for the presence of an organism in a sample. Second, we present a means to compare community structure across a collection of many samples and develop direct associations between the abundance of certain organisms and sample metadata. Third, we apply new tools to analyze the phylogenetic diversity of microbial communities and again demonstrate how this can be associated to sample metadata.
These analyses are implemented in an open source software pipeline called PhyloSift. As a pipeline, PhyloSift incorporates several other programs including LAST, HMMER, and pplacer to automate phylogenetic analysis of protein coding and RNA sequences in metagenomic datasets generated by modern sequencing platforms (e.g., Illumina, 454).
Figure 1 shows the general outline of the workflow.
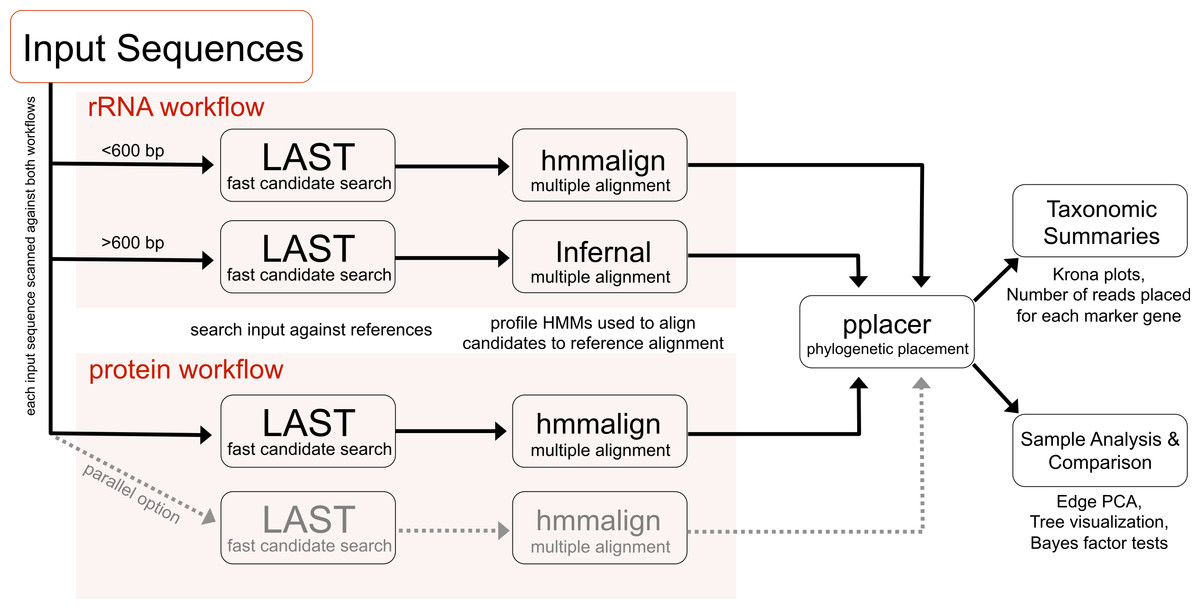 |
| Figure 1 showing the Phylosift workflow. |
The workflow follows a series of steps including
- Sequence identity search
- Alignment to reference multiple alignment
- Placement on a phylogenetic reference tree
- Visual presentation of taxonomic summary
- Comparison among samples (e.g., using Edge PCA)
In addition, there is a workflow for updating the database behind Phylosift which includes
It also provides Krona based output visualization of the taxonomic composition of a sample.

Anyway, more on Phylosift later. Just thought I would get some out here on the blog. Thanks to Aaron Darling, Holly Bik, Guillaume Jospin, Eric Lowe and Erick Matsen for all their hard work on this. And thanks to the Department of Homeland Security for supporting the work.
For more about Phylosift see
- Acquiring new genome data
- Gene family search and alignment workflow on each genome
- Phylogenetic inference and pruning
- Selection of representatives for similarity search
- Taxonomic reconciliation
 |
| Figure 2. Comparison of QIIME PCA and edge PCA analysis of human fecal samples. |
 |
| Figure 3: Lineages contributing variation in human fecal sample community structure. (Analyzed using EDGE PCA) |
It also provides Krona based output visualization of the taxonomic composition of a sample.
For more about Phylosift see


No comments:
Post a Comment