We’d like to first thank Jon for the opportunity to discuss our work in this forum. We recently published a study investigating direct functional annotation of short metagenomic reads that stemmed from protocol development for our lab. Jon invited us to write a blog post on the subject, and we thought it would be a great venue to discuss some practical applications of our work and to share with the research community the motivation for our study and how it came about.
Our lab, the Borenstein Lab at the University of Washington, is broadly interested in metabolic modeling of the human microbiome (see, for example our Metagenomic Systems Biology approach) and in the development of novel computational methods for analyzing functional metagenomic data (see, for example, Metagenomic Deconvolution). In this capacity, we often perform large-scale analysis of publicly available metagenomic datasets as well as collaborate with experimental labs to analyze new metagenomic datasets, and accordingly we have developed extensive expertise in performing functional, community-level annotation of metagenomic samples. We focused primarily on protocols that derive functional profiles directly from short sequencing reads (e.g., by mapping the short reads to a collection of annotated genes), as such protocols provide gene abundance profiles that are relatively unbiased by species abundance in the sample or by the availability of closely-related reference genomes. Such functional annotation protocols are extremely common in the literature and are essential when approaching metagenomics from a gene-centric point of view, where the goal is to describe the community as a whole.
However, when we began to design our in-house annotation pipeline, we pored over the literature and realized that each research group and each metagenomic study applied a slightly different approach to functional annotation. When we implemented and evaluated these methods in the lab, we also discovered that the functional profiles obtained by the various methods often differ significantly. Discussing these findings with colleagues, some further expressed doubt that that such short sequencing reads even contained enough information to map back unambiguously to the correct function. Perhaps the whole approach was wrong!
We therefore set out to develop a set of ‘best practices’ for our lab for metagenomic sequence annotation and to prove (or disprove) quantitatively that such direct functional annotation of short reads provides a valid functional representation of the sample. We specifically decided to pursue a large-scale study, performed as rigorously as possible, taking into account both the phylogeny of the microbes in the sample and the phylogenetic coverage of the database, as well as several technical aspects of sequencing like base-calling error and read length. We have found this evaluation approach and the results we obtained quite useful for designing our lab protocols, and thought it would be helpful to share them with the wider metagenomics and microbiome research community. The result is our recent paper in PLoS One, Comparative Analysis of Functional Metagenomic Annotation and the Mappability of Short Reads.
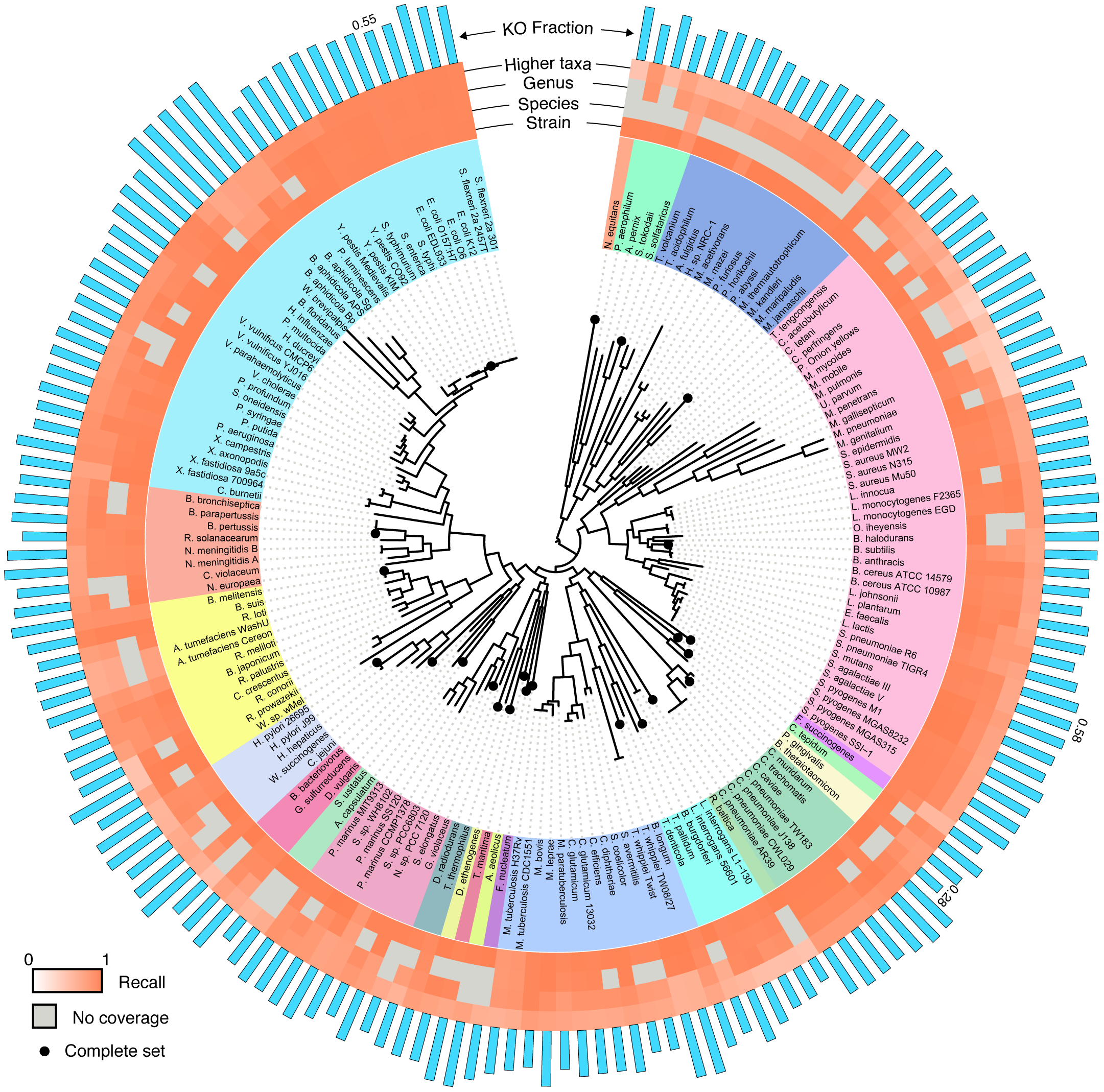 |
| The performance of BLAST-based annotation of short reads across the bacterial and archaeal tree of life using the 'top gene' protocol. See the manuscript for full details. Figure and text adapted from: Carr R, Borenstein E (2014) Comparative Analysis of Functional Metagenomic Annotation and the Mappability of Short Reads. PLoS ONE 9(8): e105776 |


